
Summary table for iLocus paper
Tonight I created the following table for my iLocus paper.
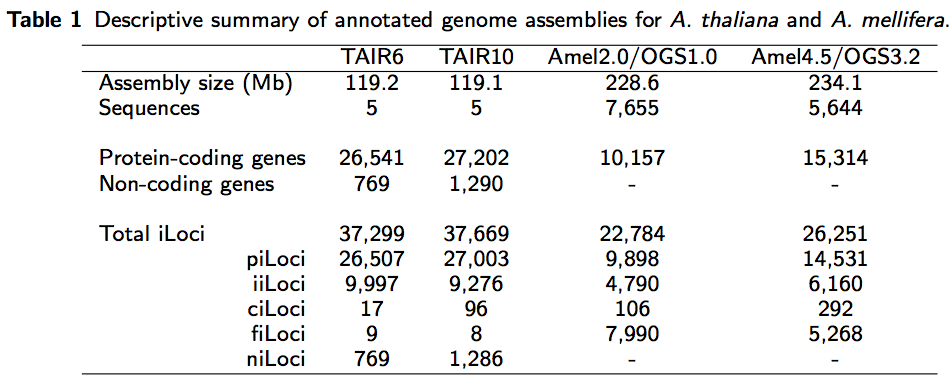
The purpose of this notebook entry is to document the commands used to populate the table while everything is still fresh in my head. The annotation files were created using GenHub v0.3.1.
genhub-build.py --workdir=species \
--genome=Am10,Am32,Att6,Atha \
--numprocs=4 \
download format prepare
In [1]:
# The input files
annotfiles="species/Att6/Att6.iloci.gff3 species/Atha/Atha.iloci.gff3
species/Am10/Am10.iloci.gff3 species/Am32/Am32.iloci.gff3"
In [2]:
# First row of the table: genome size
for file in $annotfiles; do
grep '^##sequence-region' $file | awk '{ sum += $4 } END { print sum }'
done
In [3]:
# Second row: sequence count
for file in $annotfiles; do
grep -c '^##sequence-region' $file
done
In [4]:
# Third row: gene count
for file in $annotfiles; do
grep $'\tmRNA\t' $file \
| perl -ne 'm/Parent=([^;\n]+)/ and print "$1\n"' \
| sort -u \
| wc -l \
| tr -d ' '
done
In [5]:
# Determine all annotated feature types so we can test for all
# ncRNA types.
cat $annotfiles | grep -v '^#' | cut -f 3 | sort -u
In [6]:
# Fourth row: non-coding gene count
for file in $annotfiles; do
grep -e $'\tmiRNA\t' -e $'\tncRNA\t' -e $'\trRNA\t' -e $'\tsnRNA\t' \
-e $'\tsnoRNA\t' -e $'\ttRNA\t' -e $'\ttranscript\t' $file \
| perl -ne 'm/Parent=([^;\n]+)/ and print "$1\n"' \
| sort -u \
| wc -l \
| tr -d ' '
done
In [7]:
# Fifth row: total iLocus count
for file in $annotfiles; do
grep -c $'\tlocus\t' $file
done
In [8]:
# Final rows: iLocus count breakdown
for file in $annotfiles; do
echo ""
echo $file
for type in piLocus iiLocus ciLocus fiLocus niLocus; do
echo -n "$type: "
grep -c iLocus_type=$type $file
done
done